research
Human immune systems are multicellular sensory systems shaped by
genes, development and the environment. Our past experiences with a vast
universe of molecular threats give us all unique immunological
“fingerprints”, even between genetically identical twins. I explore the
rules behind how that individuality can impact clinical outcomes in
health and immune mediated disease.
During my PhD at Cambridge with Ken Smith and at NIH with John Tsang, I focused on uncovering baseline “set-points” and dynamic response states that forecast how patients will react to vaccination, infection, and cancer immunotherapy.
Methodologically, my main approaches include application of statistical modeling and machine learning to highly multiplexed measurements such as single cell multi-omics, CyTOF, and proteomics data to longitudinal human cohorts.
As a resident in Laboratory Medicine, I’m working on building clinically deployable tests that read immune state and inform same-day treatment decisions for patients across medical specialties.
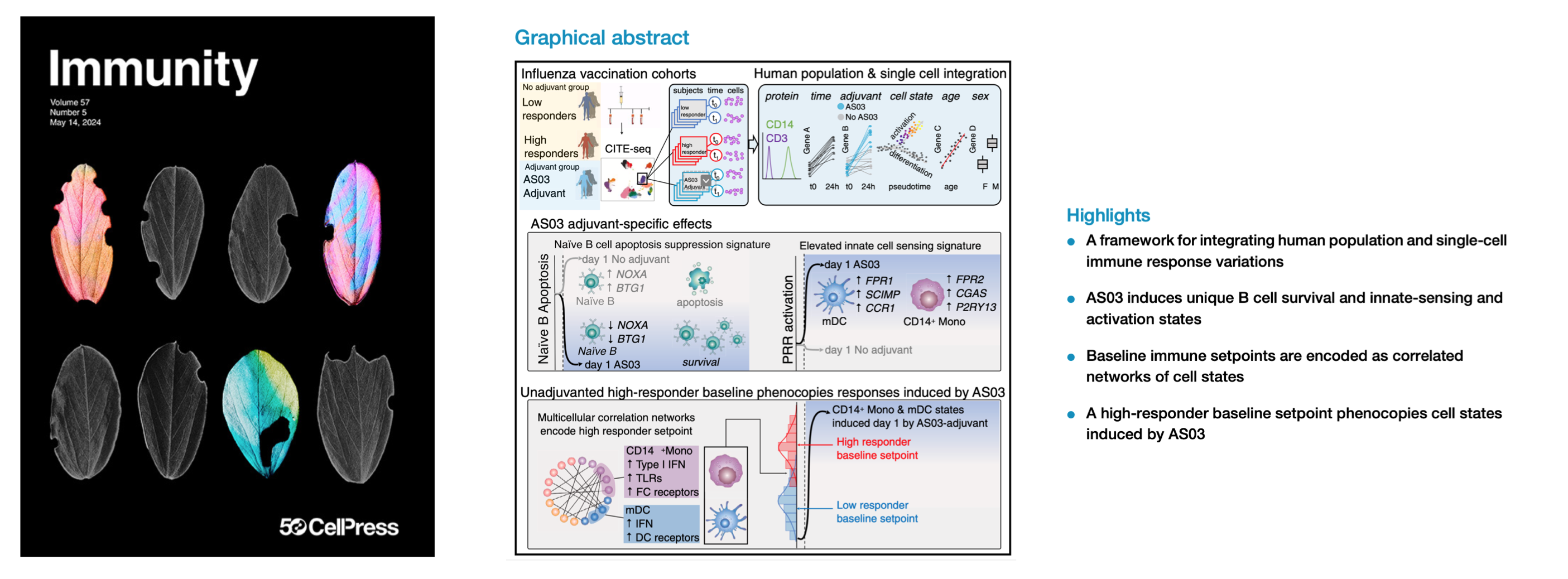
Integrating population and single-cell variations in vaccine responses identifies a naturally adjuvanted human immune setpoint
Immunity (2024) Multimodal single-cell profiling methods can capture immune cell variations unfolding over time at the molecular, cellular, and population levels. Transforming these data into biological insights remains challenging. Here, we introduce a framework to integrate variations at the human population and single-cell levels in vaccination responses. Comparing responses following AS03-adjuvanted versus unadjuvanted influenza vaccines with CITE-seq revealed AS03-specific early (day 1) response phenotypes, including a B cell signature of elevated germinal center competition. A correlated network of cell-type-specific transcriptional states defined the baseline immune status associated with high antibody responders to the unadjuvanted vaccine. Certain innate subsets in the network appeared ‘‘naturally adjuvanted,’’ with transcriptional states resembling those induced uniquely by AS03-adjuvanted vaccination. Consistently, CD14+ monocytes from high responders at baseline had elevated phospho-signaling responses to lipopolysaccharide stimulation. Our findings link baseline immune setpoints to early vaccine responses, with positive implications for adjuvant development and immune response engineering.

Normalizing and denoising protein expression data from droplet-based single cell profiling
Nature Communications (2022) Multimodal single-cell profiling methods that measure protein expression with oligo-conjugated antibodies hold promise for comprehensive dissection of cellular heterogeneity, yet the resulting protein counts have substantial technical noise that can mask biological variations. Here we integrate experiments and computational analyses to reveal two major noise sources and develop a method called “dsb” (denoised and scaled by background) to normalize and denoise droplet-based protein expression data. We discover that protein-specific noise originates from unbound antibodies encapsulated during droplet generation; this noise can thus be accurately estimated and corrected by utilizing protein levels in empty droplets. We also find that isotype control antibodies and the background protein population average in each cell exhibit significant correlations across single cells, we thus use their shared variance to correct for cell-to-cell technical noise in each cell. We validate these findings by analyzing the performance of dsb in eight independent datasets spanning multiple technologies, including CITE-seq, ASAP-seq, and TEA-seq. Compared to existing normalization methods, our approach improves downstream analyses by better unmasking biologically meaningful cell populations. Our method is available as an open-source R package that interfaces easily with existing single cell software platforms such as Seurat, Bioconductor, and Scanpy and can be accessed at “dsb [https://cran.r-project.org/package=dsb]”.

Contrasting autoimmune and treatment effects reveals baseline set points of immune toxicity following checkpoint inhibitor treatment
BioRxiv (2022) Immune checkpoint inhibitors (ICIs) have changed the cancer treatment landscape, but severe immune-related adverse events (irAEs) can trigger life-threatening autoimmunity or treatment discontinuation. Uncovering immune phenotypes associated specifically with irAEs but not antitumor immunity could help mitigate treatment discontinuation and improve clinical outcomes. We carried out simultaneous transcriptome and surface protein profiling of blood immune cells from thymic cancer patients before and after treatment with the anti-PD-L1 antibody avelumab. All patients had antitumor responses, yet a subset developed severe myositis. We developed an analytical framework to disentangle phenotypes linked to treatment responses versus irAEs. Our approach identified a temporally stable, pre-treatment immune set point associated with irAEs, but not anti-tumor effects, consisting of correlated innate and adaptive cell phenotypes. These phenotypes included potentially actionable genes downstream of mTOR in T-cell subsets. Elevated expression of gene signatures reflecting this irAE-associated setpoint at baseline in other types of cancer predicted the development of irAEs after treatment with different checkpoint inhibitors. Together our findings indicate a generalizable baseline immune setpoint linked to the development of irAEs after ICI treatment and raise the prospect of therapeutically dampening autoimmunity while sparing antitumor activity in the setting of checkpoint inhibition.